# Infection
* Common cold
* Flu
* Fever - headaches are common with fever from any type of infection
* Ear infection
* Tooth infection (type of Dental conditions)
* Sinus infection
* Pneumonia
* Measles
* Mumps
* Tonsillitis
* Sinus blockage
* Coughing - too much coughing can cause a form of traction headache.
# Various possible types of headache:
* Migraine
* Cluster headache
* Tension headache
* See also types of headache
# Lifestyle causes
* Hangover
* Excessive alcohol
* Stress
* Fatigue
* Tension
* Tiredness
* Excessive smoking
# Dyspepsia
# Eye conditions
* Glaucoma
* Eyestrain
# Medical procedures
* Spinal tap treatment
* Epidural - this anaesthetic procedure (common for childbirth) can occasionally cause damage to the spinal area and cause headache.
# Systemic or metabolic conditions
* Hypertension
* Thyroid disease
* Anemia
* Kidney failure (type of Kidney disease)
* Uremia
* Lead poisoning - African Folk Remedies - Kohl - headache
* Various toxins - see toxin causes of headache
# Brain or head conditions
* Meningitis
* Encephalitis
* Head injury
* Brain injury
* Mild traumatic brain injury
* Concussion
* Temporal arteritis
* Heatstroke
* Sunstroke
* Blood clots - in the brain, these can cause a stroke.
* Brain aneurysm
* Subdural hematoma
* Stroke
* Transient ischemic attacks
* Subarachnoid hemorrhage
* Brain tumor
* Benign intracranial hypertension
* Trigeminal neuralgia
Showing posts with label CNS. Show all posts
Showing posts with label CNS. Show all posts
Myasthenia Gravis and Lambert Eaton
Myasthenia gravi s
s
Ptosis of the left eye. Tensilon test
Imaging
 s
sMyasthenia gravis (literally "serious muscle-weakness") is a neuromuscular disease leading to fluctuating muscle weakness and fatiguability. It is an autoimmune disorder, in which weakness is caused by circulating antibodies that block acetylcholine receptors at the post-synaptic neuromuscular junction, inhibiting the stimulative effect of the neurotransmitter acetylcholine. Myasthenia is treated medically with cholinesterase inhibitors or immunosuppressants, and, in selected cases, thymectomy. At 200–400 cases per million it is one of the less common autoimmune disorders.
Signs and symptoms
Ptosis of the left eye.
The hallmark of myasthenia gravis is fatiguability. Muscles become progressively weaker during periods of activity and improve after periods of rest. Muscles that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are especially susceptible. The muscles that control breathing and neck and limb movements can also be affected. Often the physical examination is within normal limits.[3]
The onset of the disorder can be sudden. Often symptoms are intermittent. The diagnosis of myasthenia gravis may be delayed if the symptoms are subtle or variable.
In most cases, the first noticeable symptom is weakness of the eye muscles. In others, difficulty in swallowing and slurred speech may be the first signs. The degree of muscle weakness involved in MG varies greatly among patients, ranging from a localized form, limited to eye muscles (ocular myasthenia), to a severe or generalized form in which many muscles - sometimes including those that control breathing - are affected. Symptoms, which vary in type and severity, may include asymmetrical ptosis (a drooping of one or both eyelids), diplopia (double vision) due to weakness of the muscles that control eye movements, unstable or waddling gait, weakness in arms, hands, fingers, legs, and neck, a change in facial expression, dysphagia (difficulty in swallowing), shortness of breath and dysarthria (impaired speech, often nasal due to weakness of the velar muscles).
In myasthenic crisis a paralysis of the respiratory muscles occurs, necessitating assisted ventilation to sustain life. In patients whose respiratory muscles are already weak, crises may be triggered by infection, fever, an adverse reaction to medication, or emotional stress.[4] Since the heart muscle is stimulated differently, it is never affected by MG.
DiagnosisMyasthenia can be a difficult diagnosis, as the symptoms can be subtle and hard to distinguish from both normal variants and other neurological disorders. A thorough physical examination can reveal easy fatiguability, with the weakness improving after rest and worsening again on repeat of the exertion testing. Applying ice to weak muscle groups characteristically leads to improvement in strength of those muscles. Additional tests are often performed, as mentioned below. Furthermore, a good response to medication can also be considered a sign of autoimmune pathology.
Physical examination
Muscle fatigability can be tested for many muscles.A thorough investigation includes:
- looking upward and sidewards for 30 seconds: ptosis and diplopia.
- looking at the feet while lying on the back for 60 seconds
- keeping the arms stretched forward for 60 seconds
- 10 deep knee bends
- walking 30 steps on both the toes and the heels
- 5 situps, lying down and sitting up completely
- "Peek sign": after complete initial apposition of the lid margins, they quickly (within 30 seconds) start to separate and the sclera starts to show
Blood tests
If the diagnosis is suspected, serology can be performed in a blood test to identify certain antibodies:
- One test is for antibodies against the acetylcholine receptor. The test has a reasonable sensitivity of 80–96%, but in MG limited to the eye muscles (ocular myasthenia) the test may be negative in up to 50% of the cases.
- A proportion of the patients without antibodies against the acetylcholine receptor have antibodies against the MuSK protein.
- In specific situations (decreased reflexes which increase on facilitation, co-existing autonomic features, suspected presence of neoplasm, especially of the lung, presence of increment or facilitation on repetitive EMG testing) testing is performed for Lambert-Eaton syndrome, in which other antibodies (against a voltage-gated calcium channel) can be found.
Edrophonium test
Tensilon test

Imaging
A chest CT-scan showing a thymoma (red circle).
A chest X-ray is frequently performed; it may point towards alternative diagnoses (e.g. Lambert-Eaton due to a lung tumor) and comorbidity. It may also identify widening of the mediastinum suggestive of thymoma, but computed tomography (CT) or magnetic resonance imaging (MRI) are more sensitive ways to identify thymomas, and are generally done for this reason.
Pulmonary function test
Restrictive pattern on spirometry
Associations
Myasthenia Gravis is associated with various autoimmune diseases, including:
- Thyroid diseases, including Hashimoto's thyroiditis and Graves' disease
- Diabetes mellitus type 1
- Rheumatoid arthritis
- Lupus, and
- Demyelinating CNS diseases
Neurocysticercosis: Tanea Solium
Tanea Solium: Devastaiting effect.

Medical Care
Treatment of neurocysticercosis depends upon the viability of the cyst and its complications. Management includes symptomatic treatment as well as treatment directed against the parasite.
•If the parasite is dead, the treatment is directed primarily against the symptoms (eg, anticonvulsants for management of seizures). Monotherapy is usually sufficient. Duration of the treatment remains undefined, and depends neither on the type of seizure at presentation nor on other risk factors for recurrence, such as age at onset and number of seizures before diagnosis. Calcification remains an epileptogenic focus. Treating patients with viable cysts with a course of anticysticercal drugs in order to achieve better control of seizures is common practice.
•If the parasite is viable or active and the patient has vasculitis, arachnoiditis, or encephalitis, a course of steroids or immunosuppressants is recommended before the use of anticysticercal drugs. Antiparasitic treatment8 with albendazole is also useful in cysticercosis of the racemose type. If only parenchymal, subarachnoid, or spinal cysts are present without the complications mentioned, anticysticercal treatment can be considered, with the concomitant use of steroids, even in patients with massive brain infection. Reports indicate that multiple trials with anticysticercal treatment may be required for giant subarachnoid cysts.
•A recent double-blind, placebo-controlled study has shown that in patients with seizures due to viable parenchymal cysts, antiparasitic therapy decreases the burden of parasites and is safe and effective, at least in reducing the number of seizures with generalization.
Surgical Care
•In the presence of hydrocephalus due to intraventricular cyst, placement of a ventricular shunt is recommended, followed by surgical extirpation of the cyst and subsequent medical treatment.
•In cases of multiple cysts in the subarachnoid space (ie, the racemose form), surgical extirpation, on an urgent basis, is recommended.
•If the obstruction is due to arachnoiditis, placement of a ventricular shunt should be followed by administration of steroids and subsequent medical therapy.
•Because of frequent shunt dysfunctions due to entry of inflammatory tissue as well as parasitic debris inside the ventricular cavities, Sotelo designed a device that functions at a constant flow without the valvular mechanism of Pudenz-type shunts.
•Neuroendoscopy is a new tool with great potential for use in the management of ventricular cysticercosis.
•Surgical treatment in the particular case of medically refractory epilepsy due to a single lesion has been reported. Evaluation in an epilepsy center is indicated.
{kind=link}

Introduction
Neurocysticercosis (NCC) is the most common parasitic disease of the nervous system and is the main cause of acquired epilepsy in developing countries. Lately, it has also been a problem in industrialized countries because of immigration of tapeworm carriers from areas of endemic disease. Tanae Solium.
Clinical Features:
History
NCC is a pleomorphic disease, although it sometimes produces no clinical manifestation. This pleomorphism is due to variations in the locations of the lesions, the number of parasites, and the host's immune response.

•Many patients are asymptomatic; others report vague symptoms such as headache or dizziness.
•The onset of symptoms is usually subacute to chronic, with the exception of seizures, which present in an acute fashion. Patients may present with the following:
◦Epilepsy
■Epilepsy is the most common presentation (70%)
■Seizures secondary to NCC may be generalized or partial.
◦Headache
■Chronic headaches associated with nausea and vomiting (simulating migraines)
■Headaches associated with intracranial hypertension and indicative of hydrocephalus
■Headaches due to meningitis
◦Intracranial hypertension
■Most often, intracranial hypertension is due to obstruction of cerebrospinal fluid (CSF) circulation caused by basal or ventricular cysticercosis. It may also result from large cysts displacing midline structures, granular ependymitis, arachnoiditis, or the so-called cysticercotic encephalitis caused by the inflammatory response to a massive infestation of cerebral parenchyma with cysticerci.
■These patients may have seizures and deterioration of their mental status, mainly due to the host's inflammatory reaction as an exaggerated response to the massive infestation.
◦Strokes5
■Ischemic cerebrovascular complications of NCC include lacunar infarcts6 and large cerebral infarcts due to occlusion or vascular damage.
■Hemorrhage also can occur, and has been reported as a result of rupture of mycotic aneurysms of the basilar artery.
■Strokes may be responsible for the following signs and symptoms: paresis or plegias, involuntary movements, gait disturbances, or paresthesias.
◦Neuropsychiatric disturbances
■These range from poor performance on neuropsychological tests to severe dementia.
■These symptoms appear to be related more to the presence of intracranial hypertension than to the number or location of parasites in the brain.
◦Diplopia: This is a result of intracranial hypertension or arachnoiditis producing entrapment or compression of cranial nerves III, IV, or VI.
◦Hydrocephalus
■Ten to thirty percent of patients with NCC develop communicating hydrocephalus due to inflammation and fibrosis of the arachnoid villi or inflammatory reaction to the meninges and subsequent occlusion of the foramina of Luschka and Magendie.
■Noncommunicating hydrocephalus may be a consequence of intraventricular cysts.
•Other forms of neurocysticercosis
◦Ocular cysticercosis: This occurs most commonly in the subretinal space. Patients may present with decreased visual acuity, visual field defects, or monocular blindness.
◦Systemic cysticercosis: This is most common in the Asian continent. The parasites may be located in the subcutaneous tissue or muscle. Peripheral nerve involvement as well as involvement of liver or spleen have been reported.
Physical
Twenty percent or less of infected patients have abnormal neurological findings. Physical findings will depend on where the cyst is located in the nervous system and include the following:
•Cognitive decline
•Dysarthria
•Extraocular movement palsy or paresis
•Hemiparesis or hemiplegia, which may be related to stroke, or Todd paralysis
•Hemisensory loss
•Movement disorders
•Hyper/hyporeflexia
•Gait disturbances
•Meningeal signs
Causes
NCC can be acquired via fecal-oral contact with carriers of the adult tapeworm. This usually indicates the presence of a tapeworm carrier in the immediate environment (ie, household) or by accidental ingestion of contaminated food. Cases of autoingestion, in which persons with teniasis may ingest the eggs of T solium into their intestine, have been reported.
Laboratory Studies
•CSF analysis
◦Analysis of the CSF is indicated in every patient presenting with new-onset seizures or neurological deficit in whom neuroimaging shows a solitary lesion but does not offer a definitive diagnosis.
Eosinophilia in the CSF suggests neurocysticercosis (NCC); however, eosinophils also are elevated in neurosyphilis and tuberculosis of the CNS.
•Stool examination
◦Taeniasis and NCC coexist in 10-15% of patients with NCC. A recent study found that intestinal taeniasis is very common in patients with massive infestation with cysticerci but without cysticercotic encephalitis.
◦Tapeworm carriers may be identified by examining the stool of the relatives of a patient with cysticercosis encephalitis.
•Immunological tests
◦Enzyme-linked immunosorbent assay (ELISA) is the most widely used test of CSF; it has a sensitivity of 50% and a specificity of 65% for NCC.
Imaging Studies
•CT scan
•MRI
considering biopsy.
Neurocysticercosis (NCC) is the most common parasitic disease of the nervous system and is the main cause of acquired epilepsy in developing countries. Lately, it has also been a problem in industrialized countries because of immigration of tapeworm carriers from areas of endemic disease. Tanae Solium.
Clinical Features:
History
NCC is a pleomorphic disease, although it sometimes produces no clinical manifestation. This pleomorphism is due to variations in the locations of the lesions, the number of parasites, and the host's immune response.

•Many patients are asymptomatic; others report vague symptoms such as headache or dizziness.
•The onset of symptoms is usually subacute to chronic, with the exception of seizures, which present in an acute fashion. Patients may present with the following:
◦Epilepsy
■Epilepsy is the most common presentation (70%)
■Seizures secondary to NCC may be generalized or partial.
◦Headache
■Chronic headaches associated with nausea and vomiting (simulating migraines)
■Headaches associated with intracranial hypertension and indicative of hydrocephalus
■Headaches due to meningitis
◦Intracranial hypertension
■Most often, intracranial hypertension is due to obstruction of cerebrospinal fluid (CSF) circulation caused by basal or ventricular cysticercosis. It may also result from large cysts displacing midline structures, granular ependymitis, arachnoiditis, or the so-called cysticercotic encephalitis caused by the inflammatory response to a massive infestation of cerebral parenchyma with cysticerci.
■These patients may have seizures and deterioration of their mental status, mainly due to the host's inflammatory reaction as an exaggerated response to the massive infestation.
◦Strokes5
■Ischemic cerebrovascular complications of NCC include lacunar infarcts6 and large cerebral infarcts due to occlusion or vascular damage.
■Hemorrhage also can occur, and has been reported as a result of rupture of mycotic aneurysms of the basilar artery.
■Strokes may be responsible for the following signs and symptoms: paresis or plegias, involuntary movements, gait disturbances, or paresthesias.
◦Neuropsychiatric disturbances
■These range from poor performance on neuropsychological tests to severe dementia.
■These symptoms appear to be related more to the presence of intracranial hypertension than to the number or location of parasites in the brain.
◦Diplopia: This is a result of intracranial hypertension or arachnoiditis producing entrapment or compression of cranial nerves III, IV, or VI.
◦Hydrocephalus
■Ten to thirty percent of patients with NCC develop communicating hydrocephalus due to inflammation and fibrosis of the arachnoid villi or inflammatory reaction to the meninges and subsequent occlusion of the foramina of Luschka and Magendie.
■Noncommunicating hydrocephalus may be a consequence of intraventricular cysts.
•Other forms of neurocysticercosis
◦Ocular cysticercosis: This occurs most commonly in the subretinal space. Patients may present with decreased visual acuity, visual field defects, or monocular blindness.
◦Systemic cysticercosis: This is most common in the Asian continent. The parasites may be located in the subcutaneous tissue or muscle. Peripheral nerve involvement as well as involvement of liver or spleen have been reported.
Physical
Twenty percent or less of infected patients have abnormal neurological findings. Physical findings will depend on where the cyst is located in the nervous system and include the following:
•Cognitive decline
•Dysarthria
•Extraocular movement palsy or paresis
•Hemiparesis or hemiplegia, which may be related to stroke, or Todd paralysis
•Hemisensory loss
•Movement disorders
•Hyper/hyporeflexia
•Gait disturbances
•Meningeal signs
Causes
NCC can be acquired via fecal-oral contact with carriers of the adult tapeworm. This usually indicates the presence of a tapeworm carrier in the immediate environment (ie, household) or by accidental ingestion of contaminated food. Cases of autoingestion, in which persons with teniasis may ingest the eggs of T solium into their intestine, have been reported.
Laboratory Studies
•CSF analysis
◦Analysis of the CSF is indicated in every patient presenting with new-onset seizures or neurological deficit in whom neuroimaging shows a solitary lesion but does not offer a definitive diagnosis.
Eosinophilia in the CSF suggests neurocysticercosis (NCC); however, eosinophils also are elevated in neurosyphilis and tuberculosis of the CNS.
•Stool examination
◦Taeniasis and NCC coexist in 10-15% of patients with NCC. A recent study found that intestinal taeniasis is very common in patients with massive infestation with cysticerci but without cysticercotic encephalitis.
◦Tapeworm carriers may be identified by examining the stool of the relatives of a patient with cysticercosis encephalitis.
•Immunological tests
◦Enzyme-linked immunosorbent assay (ELISA) is the most widely used test of CSF; it has a sensitivity of 50% and a specificity of 65% for NCC.
Imaging Studies
•CT scan

•MRI
considering biopsy.
Medical Care
Treatment of neurocysticercosis depends upon the viability of the cyst and its complications. Management includes symptomatic treatment as well as treatment directed against the parasite.
•If the parasite is dead, the treatment is directed primarily against the symptoms (eg, anticonvulsants for management of seizures). Monotherapy is usually sufficient. Duration of the treatment remains undefined, and depends neither on the type of seizure at presentation nor on other risk factors for recurrence, such as age at onset and number of seizures before diagnosis. Calcification remains an epileptogenic focus. Treating patients with viable cysts with a course of anticysticercal drugs in order to achieve better control of seizures is common practice.
•If the parasite is viable or active and the patient has vasculitis, arachnoiditis, or encephalitis, a course of steroids or immunosuppressants is recommended before the use of anticysticercal drugs. Antiparasitic treatment8 with albendazole is also useful in cysticercosis of the racemose type. If only parenchymal, subarachnoid, or spinal cysts are present without the complications mentioned, anticysticercal treatment can be considered, with the concomitant use of steroids, even in patients with massive brain infection. Reports indicate that multiple trials with anticysticercal treatment may be required for giant subarachnoid cysts.
•A recent double-blind, placebo-controlled study has shown that in patients with seizures due to viable parenchymal cysts, antiparasitic therapy decreases the burden of parasites and is safe and effective, at least in reducing the number of seizures with generalization.
Surgical Care
•In the presence of hydrocephalus due to intraventricular cyst, placement of a ventricular shunt is recommended, followed by surgical extirpation of the cyst and subsequent medical treatment.
•In cases of multiple cysts in the subarachnoid space (ie, the racemose form), surgical extirpation, on an urgent basis, is recommended.
•If the obstruction is due to arachnoiditis, placement of a ventricular shunt should be followed by administration of steroids and subsequent medical therapy.
•Because of frequent shunt dysfunctions due to entry of inflammatory tissue as well as parasitic debris inside the ventricular cavities, Sotelo designed a device that functions at a constant flow without the valvular mechanism of Pudenz-type shunts.
•Neuroendoscopy is a new tool with great potential for use in the management of ventricular cysticercosis.
•Surgical treatment in the particular case of medically refractory epilepsy due to a single lesion has been reported. Evaluation in an epilepsy center is indicated.
Alzeimer's Disease or just a poor Memroy?
Alzheimer's Disease Fact Sheet


Alzheimer’s disease (AD) is an irreversible, progressive brain disease that slowly destroys memory and thinking skills, and eventually even the ability to carry out the simplest tasks. In most people with AD, symptoms first appear after age 60.
AD is the most common cause of dementia among older people. Dementia is the loss of cognitive functioning—thinking, remembering, and reasoning—to such an extent that it interferes with a person’s daily life and activities. According to recent estimates, as many as 2.4 to 4.5 million Americans are living with AD.
AD is named after Dr. Alois Alzheimer. In 1906, Dr. Alzheimer noticed changes in the brain tissue of a woman who had died of an unusual mental illness. Her symptoms included memory loss, language problems, and unpredictable behavior. After she died, he examined her brain and found many abnormal clumps (now called amyloid plaques) and tangled bundles of fibers (now called neurofibrillary tangles). Plaques and tangles in the brain are two of the main features of AD. The third is the loss of connections between nerve cells (neurons) in the brain.
Changes in the Brain in AD
Although we still don’t know what starts the AD process, we do know that damage to the brain begins as many as 10 to 20 years before any problems are evident. Tangles begin to develop deep in the brain, in an area called the entorhinal cortex, and plaques form in other areas. As more and more plaques and tangles form in particular brain areas, healthy neurons begin to work less efficiently. Then, they lose their ability to function and communicate with each other, and eventually they die. This damaging process spreads to a nearby structure, called the hippocampus, which is essential in forming memories. As the death of neurons increases, affected brain regions begin to shrink. By the final stage of AD, damage is widespread and brain tissue has shrunk significantly.
Very Early Signs and Symptoms
Memory problems are one of the first signs of AD. Some people with memory problems have a condition called amnestic mild cognitive impairment (MCI). People with this condition have more memory problems than normal for people their age, but their symptoms are not as severe as those with AD. More people with MCI, compared with those without MCI, go on to develop AD.
Other changes may also signal the very early stages of AD. For example, recent research has found links between some movement difficulties and MCI. Researchers also have seen links between some problems with the sense of smell and cognitive problems. Brain imaging and biomarker studies of people with MCI and those with a family history of AD are beginning to detect early changes in the brain like those seen in AD. These findings will need to be confirmed by other studies but appear promising. Such findings offer hope that some day, we may have tools that could help detect AD early, track the course of the disease, and monitor response to treatments.
Mild AD
As AD progresses, memory loss continues and changes in other cognitive abilities appear. Problems can include getting lost, trouble handling money and paying bills, repeating questions, taking longer to complete normal daily tasks, poor judgment, and mood and personality changes. People often are first diagnosed in this stage.
Moderate AD
In this stage, damage occurs in areas of the brain that control language, reasoning, sensory processing, and conscious thought. Memory loss and confusion increase, and people begin to have problems recognizing family and friends. They may be unable to learn new things, carry out tasks that involve multiple steps (such as getting dressed), or cope with new situations. They may have hallucinations, delusions, and paranoia, and may behave impulsively.
Severe AD
By the final stage, plaques and tangles have spread throughout the brain and brain tissue has shrunk significantly. People with severe AD cannot communicate and are completely dependent on others for their care. Near the end, the person may be in bed most or all of the time as the body shuts down.
What Causes AD
Scientists don’t yet fully understand what causes AD, but it is clear that it develops because of a complex series of events that take place in the brain over a long period of time. It is likely that the causes include genetic, environmental, and lifestyle factors. Because people differ in their genetic make-up and lifestyle, the importance of these factors for preventing or delaying AD differs from person to person.
The Basics of AD
Scientists are conducting studies to learn more about plaques, tangles, and other features of AD. They can now visualize plaques by imaging the brains of living individuals. They are also exploring the very earliest steps in the disease process. Findings from these studies will help them understand the causes of AD.
One of the great mysteries of AD is why it largely strikes older adults. Research on how the brain changes normally with age is shedding light on this question. For example, scientists are learning how age-related changes in the brain may harm neurons and contribute to AD damage. These age-related changes include inflammation and the production of unstable molecules called free radicals.
Genetics
In a very few families, people develop AD in their 30s, 40s, and 50s. These people have a mutation, or permanent change, in one of three genes that they inherited from a parent. We know that these gene mutations cause AD in these “early-onset” familial cases.
However, most people with AD have “late-onset” AD, which usually develops after age 60. Many studies have linked a gene called APOE to late-onset AD. This gene has several forms. One of them, APOE ε4, increases a person’s risk of getting the disease. About 40 percent of all people who develop late-onset AD carry this gene. However, carrying the APOE ε4 form of the gene does not necessarily mean that a person will develop AD, and people carrying no APOE ε4 forms can also develop AD.
Lifestyle Factors
A nutritious diet, exercise, social engagement, and mentally stimulating pursuits can all help people stay healthy. New research suggests the possibility that these factors also might help to reduce the risk of cognitive decline and AD. Scientists are investigating associations between cognitive decline and heart disease, high blood pressure, diabetes, and obesity. Understanding these relationships and testing them in clinical trials will help us understand whether reducing risk factors for these diseases may help with AD as well.
How AD Is Diagnosed
AD can be definitively diagnosed only after death by linking clinical course with an examination of brain tissue and pathology in an autopsy. But doctors now have several methods and tools to help them determine fairly accurately whether a person who is having memory problems has “possible AD” (the symptoms may be due to another cause) or “probable AD” (no other cause for the symptoms can be found). To diagnose AD, doctors:
- ask questions about the person’s overall health, past medical problems, ability to carry out daily activities, and changes in behavior and personality
- conduct tests of memory, problem solving, attention, counting, and language
- carry out medical tests, such as tests of blood, urine, or spinal fluid
- perform brain scans, such as a computerized tomography (CT) scan or a magnetic resonance imaging (MRI) test
These tests may be repeated to give doctors information about how the person’s memory is changing over time.
Early diagnosis is beneficial for several reasons. Having an early diagnosis and starting treatment in the early stages of the disease can help preserve function for months to years, even though the underlying AD process cannot be changed. Having an early diagnosis also helps families plan for the future, make living arrangements, take care of financial and legal matters, and develop support networks.
In addition, an early diagnosis can provide greater opportunities for people to get involved in clinical trials. In a clinical trial, scientists test drugs or treatments to see which are most effective and for whom they work best. (See the box, below, for more information.)
How AD Is Treated
AD is a complex disease, and no single “magic bullet” is likely to prevent or cure it. That’s why current treatments focus on several different aspects, including helping people maintain mental function; managing behavioral symptoms; and slowing, delaying, or preventing AD.
Helping People with AD Maintain Mental Function
Four medications are approved by the U.S. Food and Drug Administration to treat AD. Donepezil (Aricept®), rivastigmine (Exelon®), and galantamine (Razadyne®) are used to treat mild to moderate AD (donepezil can be used for severe AD as well). Memantine (Namenda®) is used to treat moderate to severe AD. These drugs work by regulating neurotransmitters (the chemicals that transmit messages between neurons). They may help maintain thinking, memory, and speaking skills, and help with certain behavioral problems. However, these drugs don’t change the underlying disease process and may help only for a few months to a few years.
Managing Behavioral Symptoms
Common behavioral symptoms of AD include sleeplessness, agitation, wandering, anxiety, anger, and depression. Scientists are learning why these symptoms occur and are studying new treatments—drug and non-drug—to manage them. Treating behavioral symptoms often makes people with AD more comfortable and makes their care easier for caregivers.
Slowing, Delaying, or Preventing AD
AD research has developed to a point where scientists can look beyond treating symptoms to think about addressing the underlying disease process. In ongoing AD clinical trials, scientists are looking at many possible interventions, such as cardiovascular treatments, antioxidants, immunization therapy, cognitive training, and physical activity.
Supporting Families and Caregivers
Caring for a person with AD can have high physical, emotional, and financial costs. The demands of day-to-day care, changing family roles, and difficult decisions about placement in a care facility can be hard to handle. Researchers are learning a lot about AD caregiving, and studies are helping experts develop new ways to support caregivers.
Becoming well-informed about AD is one important long-term strategy. Programs that teach families about the various stages of AD and about flexible and practical strategies for dealing with difficult caregiving situations provide vital help to those who care for people with AD.
Developing good coping skills and a strong support network of family and friends also are important ways that caregivers can help themselves handle the stresses of caring for a loved one with AD. For example, staying physically active provides physical and emotional benefits. Some AD caregivers have found that participating in an AD support group is a critical lifeline. These support groups allow caregivers to find respite, express concerns, share experiences, get tips, and receive emotional comfort. The Alzheimer’s Association, Alzheimer’s Disease Centers, and many other organizations sponsor in-person and online AD support groups across the country. There are a growing number of groups for people in the early stage of AD and their families. Support networks can be especially valuable when caregivers face the difficult decision of whether and when to place a loved one in a nursing home.
Anencephaly: Newborn Deformity
ANENCEPHALY
is a neural tube defect (a disorder involving incomplete development of the brain, spinal cord, and/or their protective coverings). The neural tube is a narrow sheath that folds and closes between the 3rd and 4th weeks of pregnancy to form the brain and spinal cord of the embryo. Anencephaly occurs when the "cephalic" or head end of the neural tube fails to close, resulting in the absence of a major portion of the brain, skull, and scalp. Infants with this disorder are born without both a forebrain (the front part of the brain) and a cerebrum (the thinking and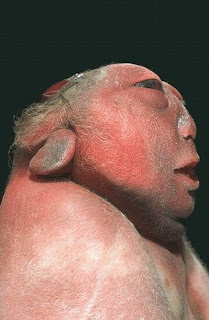 co
co ordinating area of the brain). The remaining brain tissue is often exposed--not covered by bone or skin. The infant is usually blind, deaf, unconscious, and unable to feel pain. Although some individuals
ordinating area of the brain). The remaining brain tissue is often exposed--not covered by bone or skin. The infant is usually blind, deaf, unconscious, and unable to feel pain. Although some individuals  with anencephaly may be born with a rudimentary brain stem, the lack of a functioning cerebrum permanently rules out the possibility of ever gaining consciousness. Reflex actions such as respiration (breathing) and responses to sound or touch may occur. The cause of anencephaly is unknown. Although it is believed that the mother's diet and vitamin intake may play a role, scientists believe that many other factors are also involved.
with anencephaly may be born with a rudimentary brain stem, the lack of a functioning cerebrum permanently rules out the possibility of ever gaining consciousness. Reflex actions such as respiration (breathing) and responses to sound or touch may occur. The cause of anencephaly is unknown. Although it is believed that the mother's diet and vitamin intake may play a role, scientists believe that many other factors are also involved.
is a neural tube defect (a disorder involving incomplete development of the brain, spinal cord, and/or their protective coverings). The neural tube is a narrow sheath that folds and closes between the 3rd and 4th weeks of pregnancy to form the brain and spinal cord of the embryo. Anencephaly occurs when the "cephalic" or head end of the neural tube fails to close, resulting in the absence of a major portion of the brain, skull, and scalp. Infants with this disorder are born without both a forebrain (the front part of the brain) and a cerebrum (the thinking and
 co
co ordinating area of the brain). The remaining brain tissue is often exposed--not covered by bone or skin. The infant is usually blind, deaf, unconscious, and unable to feel pain. Although some individuals
ordinating area of the brain). The remaining brain tissue is often exposed--not covered by bone or skin. The infant is usually blind, deaf, unconscious, and unable to feel pain. Although some individuals  with anencephaly may be born with a rudimentary brain stem, the lack of a functioning cerebrum permanently rules out the possibility of ever gaining consciousness. Reflex actions such as respiration (breathing) and responses to sound or touch may occur. The cause of anencephaly is unknown. Although it is believed that the mother's diet and vitamin intake may play a role, scientists believe that many other factors are also involved.
with anencephaly may be born with a rudimentary brain stem, the lack of a functioning cerebrum permanently rules out the possibility of ever gaining consciousness. Reflex actions such as respiration (breathing) and responses to sound or touch may occur. The cause of anencephaly is unknown. Although it is believed that the mother's diet and vitamin intake may play a role, scientists believe that many other factors are also involved. treatment
There is no cure or standard treatment for anencephaly. Treatment is supportive.prognosis
The prognosis for individuals with anencephaly is extremely poor. If the infant is not stillborn, then he or she will usually die within a few hours or days after birth. [Editor's Note: The unborn child may have been diagnosed as having anencephaly, but be born with a less severe form of the disease, allowing the infant to live for years or moreGullain Barre Syndrome and Miller Fisher
Guillain-Barré syndrome

Guillain-Barré syndrome (GBS) is an acute inflammatory demyelinating polyneuropathy (AIDP), an autoimmune disorder affecting the peripheral nervous system, usually triggered by an acute infectious process. It is included in the wider group of peripheral neuropathies. There are several types of GBS, but unless otherwise stated, GBS refers to the most common form, AIDP. It is frequently severe and usually exhibits as an ascending paralysis noted by weakness in the legs that spreads to the upper limbs and the face along with complete loss of deep tendon reflexes. With prompt treatment by plasmapheresis or intravenous immunoglobulins and supportive care, the majority of patients will regain full functional capacity. However, death may occur if severe pulmonary complications and dysautonomia are present.
|
|
Pathophysiology
All forms of Guillain-Barré syndrome are due to an immune response to foreign antigens (such as infectious agents) that are mistargeted at host nerve tissues instead (a form of antigenic mimicry). The targets of such immune attack are thought to be gangliosides, which are complex glycosphingolipids present in large quantities on human nerve tissues, especially in the nodes of Ranvier. An example is the GM1 ganglioside, which can be affected in as many as 20-50% of cases, especially in those preceded by Campylobacter jejuni infections. Another example is the GQ1b ganglioside, which is the target in the Miller Fisher syndrome variant
The most common antecedent infection is Campylobacter jejuni. However, 60% of cases do not have a known cause.
The end result of such autoimmune attack on the peripheral nerves is inflammation of myelin and conduction block, leading to a muscle paralysis that may be accompanied by sensory or autonomic disturbances.
Serum sickness can rarely manifest as the Guillain-Barre syndrome (GBS)
Signs and symptoms- weakness which affects the lower limbs first, and rapidly progresses in an ascending fashion.
- Frequently, the lower cranial nerves may be affected, leading to bulbar weakness, (oropharyngeal dysphagia, that is difficulty with swallowing, drooling, and/or maintaining an open airway) and respiratory difficulties.
- Most patients require hospitalization and about 30% require ventilatory assistance.
- Facial weakness is also commonly a feature, but eye movement abnormalities are not commonly seen in ascending GBS, but are a prominent feature in the Miller-Fisher variant
- Sensory loss, if present, usually takes the form of loss of proprioception (position sense) and areflexia (complete loss of deep tendon reflexes), an important feature of GBS.
- Loss of pain and temperature sensation is usually mild. In fact, pain is a common symptom in GBS, presenting as deep aching pain, usually in the weakened muscles, which patients compare to the pain from overexercising.
- These pains are self-limited and should be treated with standard analgesics. Bladder dysfunction may occur in severe cases but should be transient. If severe, spinal cord disorder should be suspected.
- Fever should not be present, and if it is, another cause should be suspected.
- In severe cases of GBS, loss of autonomic function is common, manifesting as wide fluctuations in blood pressure, orthostatic hypotension, and cardiac arrhythmias.
Six different subtypes of Guillain-Barre syndrome (GBS) exist:
- Acute inflammatory demyelinating polyneuropathy (AIDP)
- Miller Fisher syndrome (MFS)
- Acute motor axonal neuropathy (AMAN)
- Acute motor sensory axonal neuropathy (AMSAN)
- Acute panautonomic neuropathy
- Bickerstaff’s brainstem encephalitis (BBE)
Diagnosis
The diagnosis of GBS usually depends on findings such as rapid development of muscle paralysis, areflexia, absence of fever, and a likely inciting event. CSF and ECD is used almost every time to verify symptoms, but because of the acute nature of the disorder, they may not become abnormal until after the first week of onset of signs and symptoms.
There currently is no cure for Guillain-Barre syndrome. However, treatments have been proven effective against this syndrome.
- CSF
- Typical CSF findings include albumino-cytological dissociation. As opposed to infectious causes, this is an elevated protein level (100 - 1000 mg/dL), without an accompanying pleocytosis (increased cell count). A sustained pleocytosis may indicate an alternative diagnosis such as infection.
Electrodiagnostics
- Electromyography (EMG) and nerve conduction study (NCS) may show prolonged distal latencies, conduction slowing, conduction block, and temporal dispersion of compound action potential in demyelinating cases. In primary axonal damage, the findings include reduced amplitude of the action potentials without conduction slowing.
Diagnostic criteria
Required
- Progressive, relatively symmetrical weakness of 2 or more limbs due to neuropathy
- Areflexia
- Disorder course <>
- Exclusion of other causes (see below)
Supportive
- relatively symmetric weakness accompanied by numbness and/or tingling
- mild sensory involvement
- facial nerve or other cranial nerve involvement
- absence of fever
- typical CSF findings obtained from lumbar puncture
- electrophysiologic evidence of demyelination from electromyogram
Treatment
Supportive care with monitoring of all vital functions is the cornerstone of successful management in the acute patient. Of greatest concern is respiratory failure due to paralysis of the diaphragm.
Early intubation should be considered in any patient with a vital capacity (VC) <20>2O, more than 30% decrease in either VC or NIF within 24 hours, rapid progression of disorder, or autonomic instability.
Once the patient is stabilized, treatment of the underlying condition should be initiated as soon as possible.
Either high-dose intravenous immunoglobulins (IVIg) at 400 mg/kg for 5 days or
plasmapheresis
Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic Lateral Sclerosis (ALS)
Pop Star Jason beckeris surviving it.

Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig's disease, is a disease that attacks the nerve cells (motor neurons) that control muscles. It gets worse over time (is progressive). Motor neurons carry messages about movement from the brain to the muscles, but in ALS the motor neurons degenerate and die; therefore, the messages to move no longer get to the muscles. When muscles aren't used for a long time, they weaken, waste away (atrophy), and twitch under the skin (fasciculate).
Eventually, all the muscles that a person can control (voluntary muscles) are affected. People with ALS lose the ability to move their arms, legs, mouth, and body. It may get to the point that the muscles used for breathing are affected, and the person might need a respirator (ventilator) in order to breathe. People with ALS can live 3 to 10 years or more after diagnosis.
Eventually, all the muscles that a person can control (voluntary muscles) are affected. People with ALS lose the ability to move their arms, legs, mouth, and body. It may get to the point that the muscles used for breathing are affected, and the person might need a respirator (ventilator) in order to breathe. People with ALS can live 3 to 10 years or more after diagnosis.
Mental changesFor a long time it was believed that ALS only affected muscles. It is now known that one-third to one-half of individuals with ALS experience some changes in thinking (cognition). The disease can also cause changes in personality and behavior.
Who gets it?
People are most commonly diagnosed with ALS between the ages of 40 and 70, but younger people can also develop it. ALS affects people all over the world, in all ethnic backgrounds. Men are affected more often than women. About 90-95% of ALS cases appear at random, meaning no one in the person's family has the disorder. In about 5-10% of cases, a family member also has the disorder.
What causes it?
The exact cause of ALS is not known. In 1991, researchers identified a link between ALS and Chromosome 21. Two years later, a particular gene, SOD1, was identified as being associated with about 20% of the inherited cases in families. SOD1 controls an enzyme that breaks down free radicals, harmful particles that attacks cells from the inside and cause their death. Since not all inherited cases are connected to this gene, and some people are the only ones in their families with ALS, other genetic causes must exist.
Symptoms and diagnosis
Usually ALS comes on slowly, starting out as weakness in one or more muscles. Only one leg or arm may be affected. People notice that they stumble, having trouble lifting things, or have trouble with using their hands. As the disease progresses, the person with ALS will not be able to stand or walk, have trouble moving around, and trouble talking and swallowing. The diagnosis of ALS is based on the symptoms and signs the physician observes, as well as tests eliminating all the other possibilities, such as multiple sclerosis, post-polio syndrome, or infectious diseases.
Treatment
There is as yet no cure for ALS. Treatments are designed to relieve the symptoms and improve the quality of life for people with the disorder. Medications can help reduce fatigue, ease muscle cramps, and lessen pain. There is also a specific medication for ALS, called Rilutek (riluzole). It does not repair the damage already done to the body, but appears to be modestly effective in prolonging the survival of people with ALS. Through physical therapy, special equipment, and speech therapy, people with ALS can remain mobile and able to communicate.
LeaRN ABOUT bECKER AT www.xyzmusic.blogspot.com
Subscribe to:
Posts (Atom)